Pharmazeutische Produkte werden unter Einhaltung strenger Wirksamkeits- und Qualitätsstandards hergestellt. Alle Qualitätsaspekte werden unter Berücksichtigung der mit der Verabreichungsmethode (Injektion, Einnahme usw.) und der Art der Herstellung (aseptisch, sterilisiert oder unter weniger kontrollierten Bedingungen) verbundenen Risiken überprüft. Die vorliegende Arbeit befasst sich mit zwei Teilen dieses Prozesses: der Qualität der Umgebung, in der das Produkt hergestellt wird, und den Normen, die die Grenzwerte für die Partikelkonzentration festlegen, die bestimmen, was eine kontrollierte Umgebung ausmacht.
In diesem Papier werden die Normen für physikalische Prüfungen (EN ISO 14644-1:2015) und die Normen, die in behördlichen Leitlinien (EU-GMP-Annex 1) gelten, untersucht.
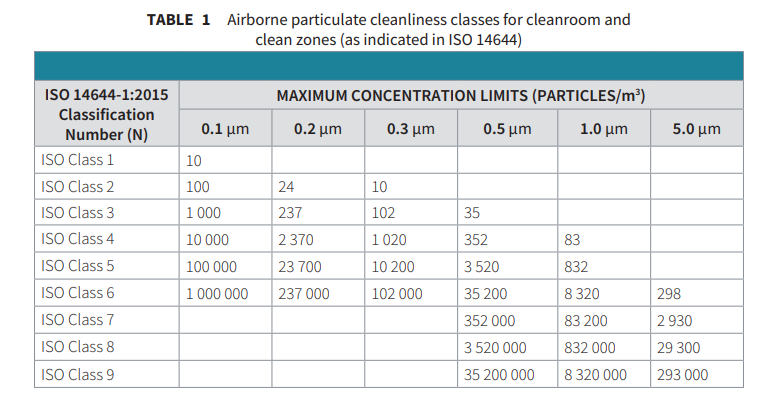
1999 wurde die neue Normenreihe ISO 14644 zur Raumklassifizierung in Kraft gesetzt. Die erste Norm war ISO 14644-1, in der die Methode zur Klassifizierung eines Raums und die maximal zulässigen Partikel innerhalb eines festgelegten Luftvolumens festgelegt wurden. Der Leser sollte beachten, dass die ISO 14644 zwar weltweit für die Klassifizierung von Reinräumen übernommen wurde, es jedoch Unterschiede bei der Routineüberwachung gibt, insbesondere zwischen der ISO 14644 und der EU- und WHO-GMP.
Der Zertifizierungszustand des Reinraums muss im Vorfeld der Prüfung definiert werden; im Rahmen der ISO 14644-1 gibt es drei Zustände:
As Built: ein fertiggestellter Raum mit allen Anschlüssen und funktionsfähig, aber ohne Produktions
Produktionsanlagen oder Personal in der Einrichtung.
At Rest: Alle Dienste sind angeschlossen, die gesamte Ausrüstung ist installiert und funktioniert in der vereinbarten Weise.
Betrieb, aber es ist kein Personal anwesend.
Operational: Die gesamte Ausrüstung ist installiert und funktioniert in einem vereinbarten Format, und eine bestimmte Anzahl
von Personal ist anwesend, das nach einem vereinbarten Verfahren arbeitet.
Füllen Sie das Formular aus, um das vollständige Dokument herunterzuladen.





